The overarching goal of our research is to improve therapies for patients with lung cancer. Our primary focus is to define the molecular underpinnings of sensitivity and resistance to targeted therapies for lung cancers with specific genetic abnormalities (EGFR mutations, ALK translocations, KRAS mutations, etc.).
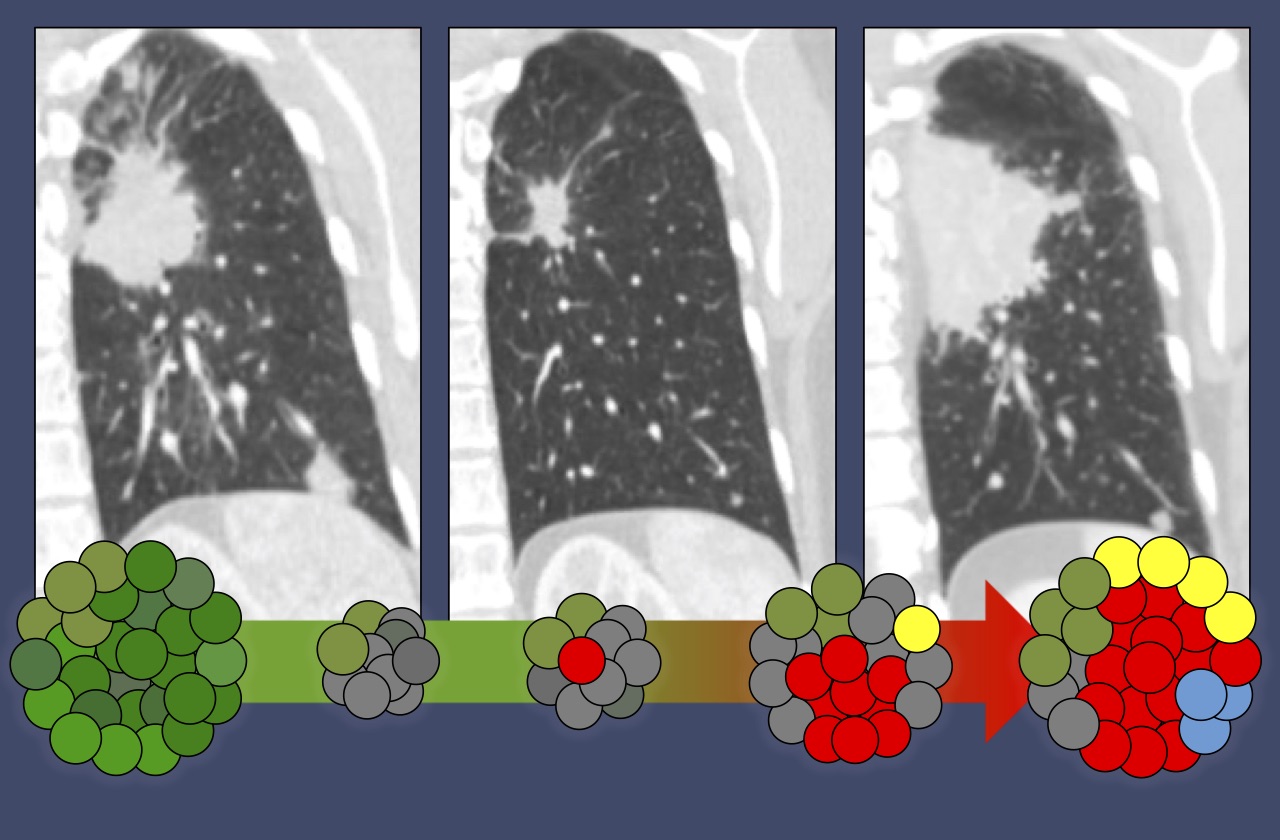
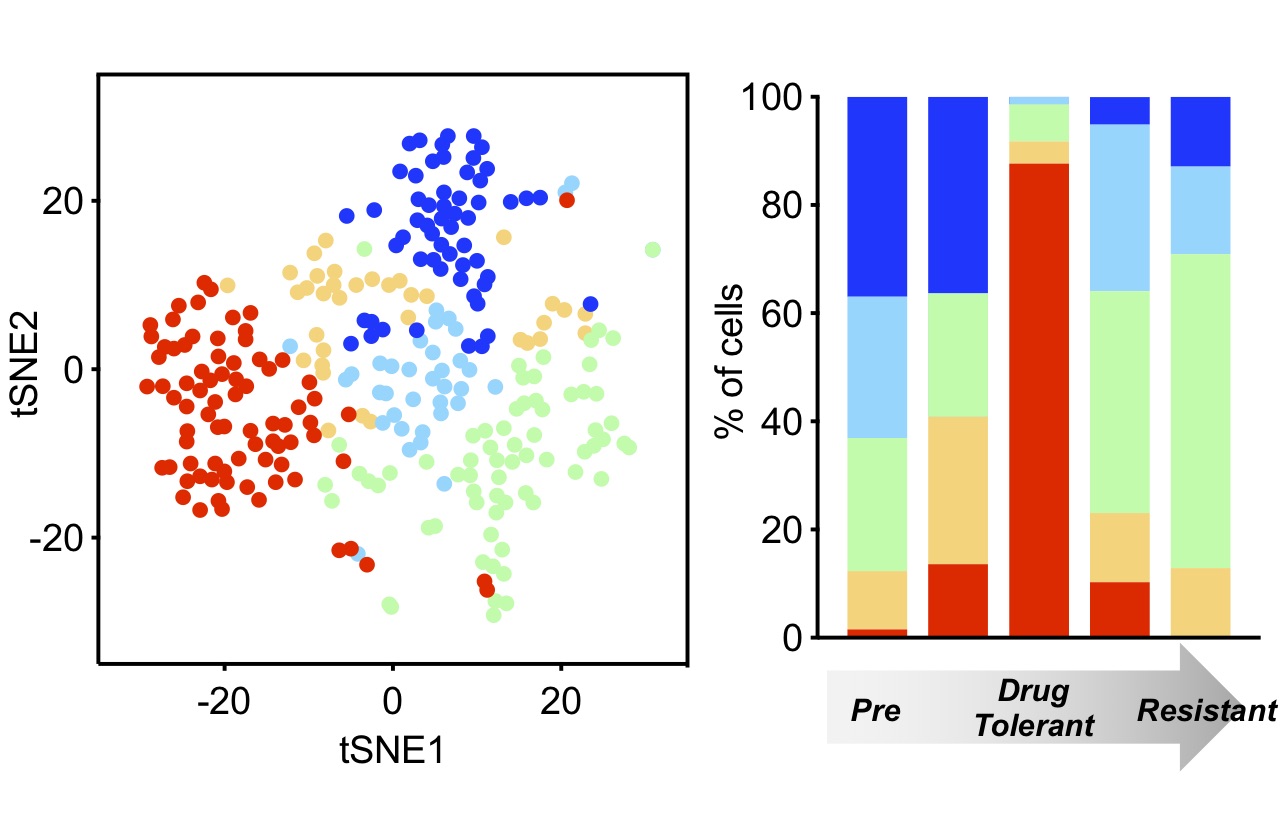
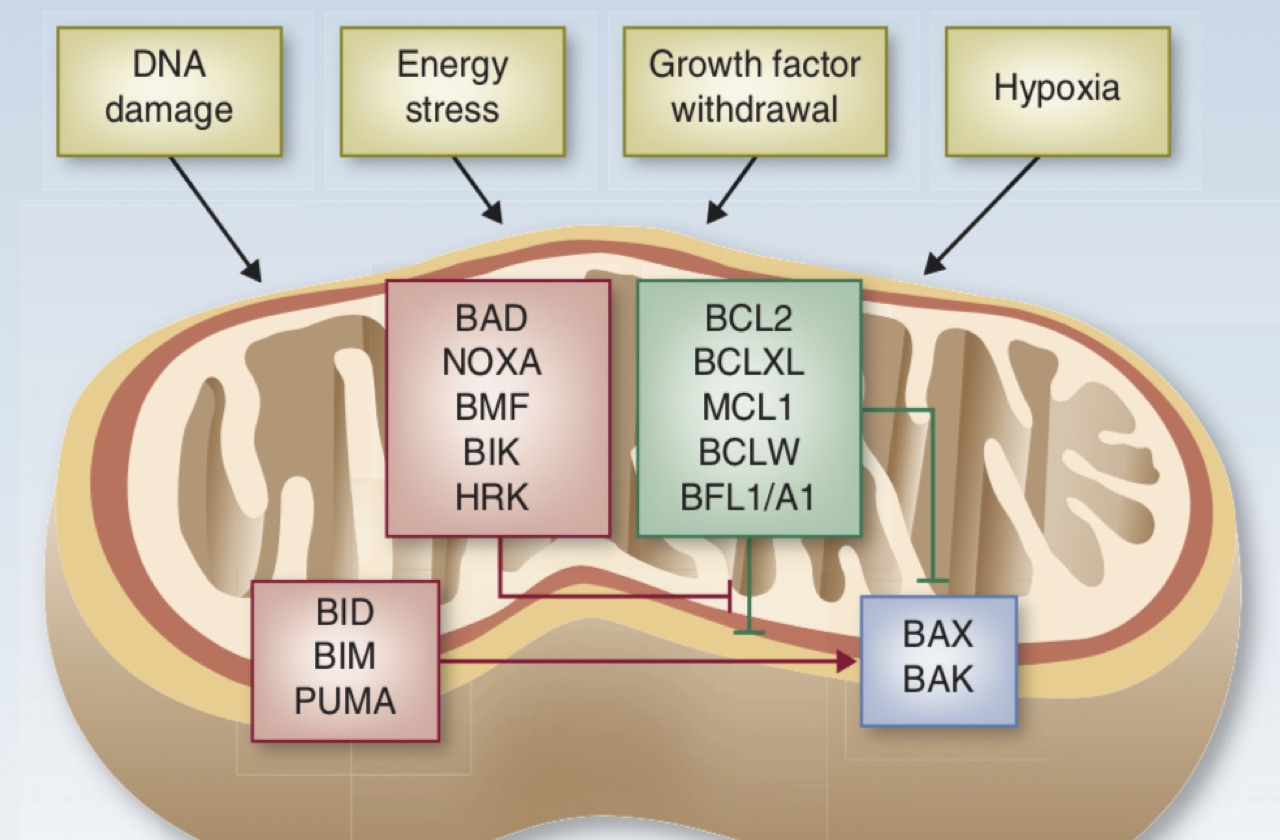
In particular, we seek to understand how kinase inhibitors modulate signaling networks that regulate cancer cell proliferation, apoptosis and genomic stability. For some lung cancer subsets, such as those with EGFR mutations and ALK translocations, effective targeted therapies have supplanted chemotherapy as the standard of care. For these patients, acquired drug resistance remains a major clinical challenge, and we are studying how molecular mechanisms of acquired resistance evolve in patients during therapy and designing therapies to overcome them. For other lung cancers, such as those with KRAS mutations, effective targeted therapies have so far remained elusive, and our efforts are focused on discovering new vulnerabilities that might be exploited. In addition, we are seeking to develop novel immunotherapy approaches for lung cancers with low mutation burdens.
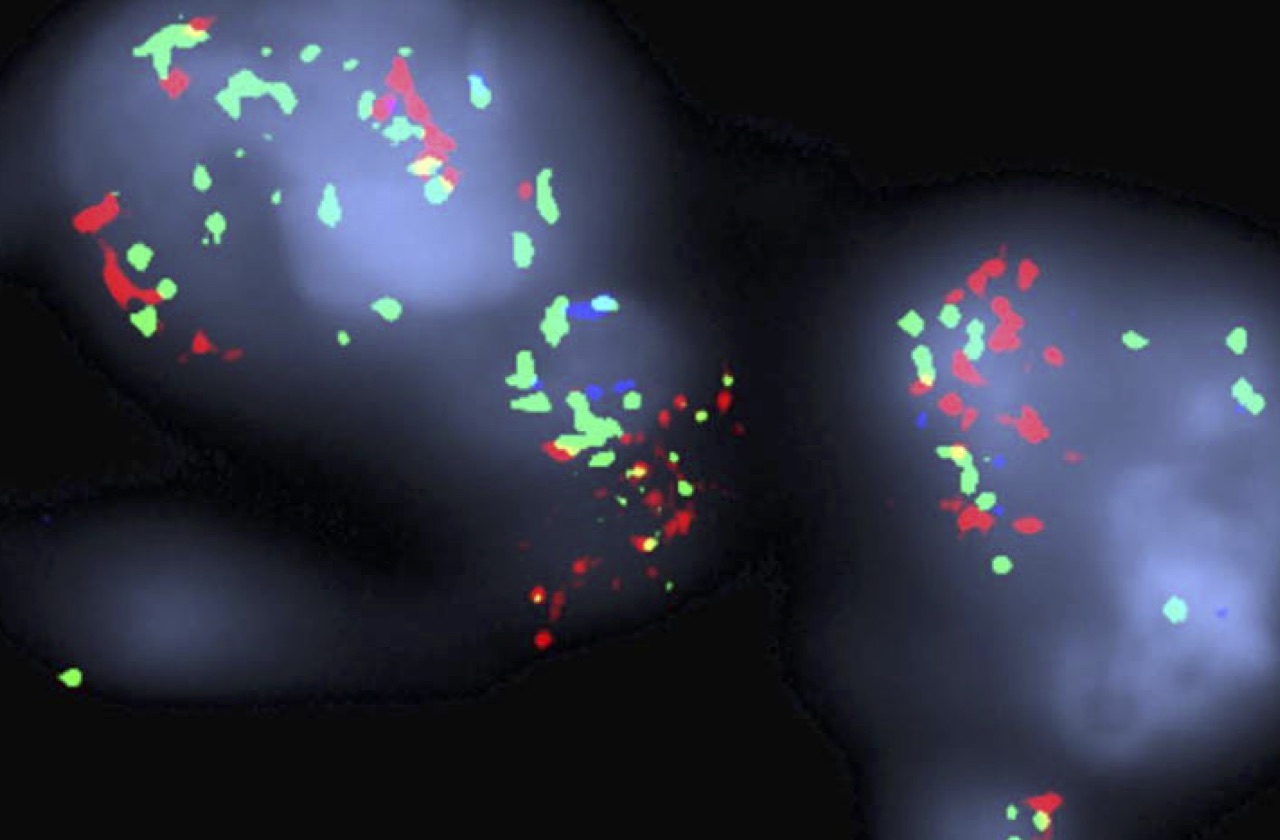
Our research is highly translational, integrating detailed mechanistic study of patient-derived cell culture and mouse (PDX) models with assessment of clinical tumor specimens, all performed in close collaboration with clinicians in the MGH Thoracic Oncology Group.